溴代烷
溴代烷,又名溴代烷烴,是有機溴化合物的一類,指的是烷烴分子中的一個或多個氫原子被溴取代的有機化合物,屬於鹵代烴的一種。通式為。而最簡單的溴代烷為一溴甲烷(甲基溴)。它們的密度大都比水大,而同一烴基的鹵代烷的密度順序為氯代烷<溴代烷<碘代烷[1]。大部分的溴代烷都不溶於水、溶於大部分的有機溶劑[2]。


溴代烷可以用作有機溶劑和有機合成的中間體(烷基化試劑[3])。但溴代烷的毒性限制了其應用範圍。而人類較常使用甲基溴做熏蒸的用途[4],但已漸漸被淘汰。
溴代烷天然存在於大自然中。據估計,海洋每年排放1到2百萬噸的溴仿和56,000噸的溴甲烷[5]。紅藻會把溴代烷和碘代烷濃縮在囊泡細胞裏,紫杉狀海門冬的揮發性精油中有95%都是鹵代烴,其中有80%都是溴仿[6] 。下圖是大自然中的有機溴化合物的例子,最左邊的一種屬於溴代烷:

左到右:溴仿、一種溴代二酚、骨螺紫、tambjamine B。

歷史
鹵代烷在很久以前已被應用,如在1847年蘇格蘭醫生詹姆斯·楊·辛普森及其朋友發現氯仿可以用作麻醉劑[7][8]。在溴代烷的結構方面,於1857年凱富勒發現碳有四價[9]、1861年布特列洛夫提出化學結構理論、1974年范霍夫和勒·貝爾提出碳的正四面體構型假說(又稱范托浩夫—勒·貝爾模型),這些都對化學家對溴代烷結構的研究起了重大的作用。在合成溴代烷的方面,在19世紀,由於有機化學蓬勃而有系統的發展,化學家們找到更多合成溴代烷的方法[10]。例如俄國化學家亞歷山大·波菲里耶維奇·鮑羅丁在1861年嘗試用乙酸銀來製備甲基溴時,發現了漢斯狄克反應。對於由烯烴合成鹵代烷的反應(烯烴與氫鹵酸的親電取代反應)的區域選擇性,在1869年,俄國化學家馬可尼可夫發現親電試劑中的正電基團(如氫)總是加在取代最少的碳原子上[11],但烯烴和溴化氫的反應卻不一定得到這個結果。直到1933年莫里斯·S·卡拉施給出了解析:過氧化物效應[12],即是在過氧化物存在下,烯烴和溴化氫的反應是反馬氏規則的,因爲反應涉及到自由基。
分類
跟其他鹵代烷一樣,溴代烷亦可分爲一級(伯)、二級(仲)、及三級(叔)溴代烷。一般來説,連著溴原子的碳連接著多少個碳,就是幾級溴代烷。
-
 一級溴代烷的結構
一級溴代烷的結構 -
 二級溴代烷的結構
二級溴代烷的結構 -
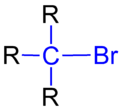 三級溴代烷的結構
三級溴代烷的結構


例如,1-溴丙烷為一級溴代烷,2-溴丙烷為二級溴代烷[13],2-溴-2-甲基丙烷為三級溴代烷。
命名
溴代烷通常以普通命名法來命名,找出相應烷烴作爲母體,將溴視作取代基,並以數字標明其位置(先寫溴,再寫烷基取代基,最後寫母體)[14]。而不少溴代烴都有俗名,例如溴仿(三溴甲烷)。同時,亦可以把烷基當作是取代基來命名,即先寫烷基再寫溴,例如乙基溴、甲基溴等[15]。
合成
烷烴的自由基取代反應
烷烴在紫外光照的條件下,可以與溴反應生成相應的溴代烴和溴化氫。反應機理有三個步驟[16]:
- 鍵引發:
- 鍵增長:
- 一、溴自由基跟一分子烷烴反應,生成溴化氫和相應的烷烴自由基。
- 二、烷烴自由基跟另一分子的溴反應,生成溴代烷和另一溴自由基。
- 鍵終止:有三個終止的反應,直到自由基消失,反應結束。
- 1.兩個溴自由基反應,生成一分子溴。
- 2.兩個烷基自由基反應,生成另一碳鏈更長的烷烴。
- 3.一個溴自由基和一個烷基自由基反應,生成溴代烷。
反應是個連鎖反應,會繼續生成二溴烷烴、三溴烷烴、四溴烷烴[18]。
烯烴、炔烴的加成反應
親電加成
烯烴和炔烴都具備π電子,可以與親電試劑發生親電加成反應。其中與溴化氫和溴的加成反應可製備溴代烷。這些反應一般遵從馬氏規則。
烯烴、炔烴與溴完全反應,分別生成1,2-二溴代烷和1,1,2,2-四溴代烷。反應跟從環鎓離子機理[19],如下圖所示。其中在溴分子接近烯烴、炔烴時,形成了誘發偶極,受到π電子的進攻,形成溴鎓離子。此溴鎓離子再受到帶負電荷的溴離子的進攻,生成產物。

烯烴、炔烴與溴化氫反應,分別生成溴代烷和1,1-溴代烷。反應遵從馬氏規則,因爲機理中涉及到碳陽離子,而由於碳陽離子會發生重排,因此有機會生成重排產物。機理是典型的親電加成反應,首先是π電子進攻溴化氫,得到質子而生成碳陽離子和溴離子。之後溴離子進攻碳陽離子,生成溴代烷。機理如下:[20] [21] [22]

自由基加成
在過氧化物的存在下,烯烴跟溴化氫的反應會是反馬氏規則的,因爲這涉及到自由基的生成[23]。例如:

醇的親核取代反應
醇可以跟氫溴酸反應,生成相應的溴代烷。但由於氫溴酸的酸性不夠,所以需要用硫酸來增強酸性。這反應以三級醇最快,二級醇次之,一級醇最慢[24]。在反應體系中加入溴化鉀,可以增加溴離子的濃度,並起了鹽析的作用,是這反應的改良[25]。
醇可以在紅磷與氫溴酸的體系中轉化為溴代烷[26]。也可以直接用三溴化磷跟醇反應,生成溴代烷。
其他
在阿佩爾反應中,用四溴化碳或溴作為鹵原子源,可由醇製備出溴代烷[27]。伯醇、仲醇和多數叔醇都能順利發生反應。
在漢斯狄克反應中,羧酸的銀鹽在無水的惰性溶劑如四氯化碳中與一分子溴回流,失去二氧化碳並形成比羧酸少一個碳的溴代烷[28][29][30][31] 。

漢斯狄克反應屬於自由基機理。首先羧酸銀與溴 (1) 快速反應而生成中間體 (2)。中間體在受熱的情況下均裂為酰氧基自由基和溴自由基 (3),然後 RCOO· 分解放出二氧化碳,生成烷基自由基 R·,並重新與溴自由基結合為溴代烴 (5)。

同時,可以透過鹵仿反應,將含乙酰基的有機化合物轉變成溴仿[32]。

Haloform reaction scheme

首先,羰基的α-氫在鹼性環境之下不斷被溴取代[33],取代的過程重複三次,直到所有的氫原子都被換成溴原子。然後,氫氧根離子進攻羰基碳, 離子離去。 離子被碳上三個吸電子基團(溴)所穩定。最後一步為酸鹼反應, 從羧酸取得一質子,生成羧酸根離子和溴仿。其後羧酸根離子可以得到質子,重新生成羧酸。所生成的羧酸比原本的羰基化合物少一個碳[34]。反應的機理如下:



反應
親核取代反應
溴的電負性比碳的強,因此碳原子上有部分正電荷,可以受親核試劑的進攻。而溴是一個挺好的離去基團,使溴代烷相對上比較容易發生親核取代反應,可以被氫氧根離子等各種親核試劑進攻。例如,溴乙烷和氫氧根離子發生雙分子親核取代反應,經過五配位的過渡態後生成乙醇。

又如溴乙烷與乙酸鈉反應,作爲親核試劑的乙酸根離子(乙酰氧基負離子)進攻,溴以溴離子的形式離去,生成乙酸乙酯:

由於位阻效應較小和所生成的一級碳陽離子不穩定,一級溴代烷通常以雙分子親核取代反應的反應機理進行反應。而三級溴代烷因爲位阻較大,而且能生成較穩定的碳陽離子,所以通常以單分子親核取代反應的機理而進行反應[35][36]。
值得注意的是溴代烷和兩位負離子的反應。以亞硝酸根負離子做例子:一級溴代烷與其發生SN2反應,由於氮的親核性強,產物為硝基烷。但如果是二級、三級溴代烷與其作SN1反應,由於碳陽離子跟亞硝酸根負離子上負電荷較爲集中的氧有正負相吸,因此產物主要為亞硝酸酯[37]。
威廉姆遜合成反應
醇羥基在鹼性條件下去質子化,形成醇負離子,進攻溴代烷中帶部分正電荷的碳。溴是離去基團,以溴離子的形式離開,形成醚鍵[38]。這是一個合成醚的重要反應。反應機理為雙分子親核取代反應,如下:

科爾貝腈合成
氰離子進攻溴代烷,生成比原本溴代烷多一個碳的腈[39]。所生成的腈可以進一步反應為其他有機化合物(如水解成羧酸[40]、催化加氫合成胺[41][42]),因此這反應在有機合成上非常重要。

例如1-溴丁烷和氰化鈉作用得到正戊腈和大約1%的異腈,此反應在二甲亞碸中進行得到93%產率[43]。
加布里爾伯胺合成反應
鄰苯二甲酰亞胺的鈉鹽或鉀鹽與一級溴代烷發生親核取代反應,生成烷基鄰苯二甲酰亞胺,再轉換成一級胺[44][45][46][47]和副產物鄰苯二甲酰肼[48]。此反應無法製備二級胺(仲胺),因爲鄰苯二甲酰亞胺的氮上只有一個氫原子,只能引入一個烷基。反應機理如下:

烷基化
溴的電負性比碳大 (2.9 > 2.5)。因此,碳-溴鍵是極性共價鍵,其中的碳為親電性的,可被親核試劑進攻,從而增長碳鏈,因此溴代烷可以用作烷基化試劑。
酯、酮等含羰基的化合物可以在強鹼的作用下失去質子,生成烯醇負離子[49][50][51][52]。而烯醇負離子可被視作親核試劑,進攻溴代烷,生成α-烴基化的羰基化合物,這反應速率最快的為一級溴代烷和甲基溴[53]。

烯醇負離子的共振式

溴代烷更可以跟乙炔鈉等炔化物反應,生成內部炔烴,加長碳鏈。除此之外,溴代烷可以參與乙酰乙酸酯合成[54]、丙二酸酯合成[55][56]等反應,這些反應的過程亦涉及到烷基化。以下是1,4-二溴丁烷進行分子內丙二酸酯合成(珀金脂環化合物合成)生成環烷羧酸的例子:
.png)
消除反應
溴代烷可以發生消除反應,生成烯烴。而產物主要爲柴瑟夫產物,即比較穩定的烯烴[57]。
雙分子消除反應
溴代烷在強鹼的作用下失去一分子的溴化氫,生成烯烴。離去的溴和β氫必定處於反式共平面狀態[58]。由於反應由一步完成(鹼拉走質子(進攻β-氫)時與離去基同時離去),因此雙分子消除反應與二種反應物濃度皆有關,在反應動力學上是屬於二級反應。若果使用越強的鹼,反應越快進行。1-溴-2-甲基丙烷和乙醇鉀的反應機理如下:

Scheme 1. E2 reaction mechanism

單分子消除反應
三級溴代烷在沒有鹼(溶劑解)的消除反應通常為單分子消除反應。第一步是溴代烷的溴以離去基團的形式離去,生成碳正離子。這一步的速率較慢,因此是速率控制步驟。第二步是溶劑分子奪取碳正離子β-氫,生成烯烴。由於反應的速率控制步驟是溴離去的一步,只與一個底物分子有關,因此反應是單分子過程,在反應動力學上是一級反應[42]。而由於反應中的碳陽離子有機會發生重排,因此單分子消除反應有機會生成重排產物。 以2-溴-2-甲基丙烷的單分子消除反應作例,反應機理如下:

單分子消除反應

與金屬的反應
格氏試劑的製備

格氏試劑一般由溴代烷與金屬鎂在無水乙醚或四氫呋喃中直接反應而製備的[59]。由於格氏試劑非常活潑,因此製備時不能接觸水、二氧化碳,或其他含有活潑氫的化合物(如醇),需要在惰性氣體保護下製備格氏試劑。
格氏試劑是一個很好的親核試劑,可以與不同的化合物反應(如醛、酮、二氧化碳等),增長有機化合物的碳鏈,在有機合成中非常重要[60]。
有機鋰試劑的製備

而由於有機鋰試劑比格氏試劑更爲活潑,所以製備的條件也要更爲嚴格。
而二分子烴基鋰與一分子鹵化亞銅可生成二烴基銅(吉爾曼試劑)[62]:


偶聯反應

吉爾曼試劑與有機鹵化物的化學反應方程式

在武茲反應裏,兩分子的溴代烴與鈉反應,生成更長的碳鏈[64]:

其他的金屬(如銀等)都可參與反應[65]。鄰二溴化物發生武茲反應,會生成烯烴;偕二溴化物反應,會生成炔烴;而(1,3), (1,4), (1,5),或(1,6)二溴化物發生這個反應,則會生成環狀化合物[66]。而此反應雖然只限制於對稱烷烴的合成,但其用處卻是無可否認的,特別是在關很多小分子環(尤其是三元環)的時候相當有用。 比如二環丁烷就是通過1-溴-3-氯環丁烷和鈉來合成的:

還原反應
溴代烷可以被氫化鋁鋰還原成相應的烷烴[68][69],反應一般在乙醚或四氫呋喃中進行。例如溴辛烷能被還原成辛烷,產率為72%[70]。溴代烷的反應比碘代烷的慢(碘代烷的反應最快)。此反應中以還原一級溴代烷為最適合,所得產物發生構型轉化,因此認為該反應是雙分子親核取代反應的機理。二級溴代烷也可用此法還原,三級溴代烷容易發生消除反應,不適合用這個方法。[71]
鑒定
溴代烷與碘化鈉的丙酮溶液反應會生成溴化鈉。而由於溴化鈉是不溶於丙酮的,會沉澱出來,因此可用這反應來區分溴代烷[72]。化學式如下:

例子
-
 溴甲烷
溴甲烷 -
 溴仿
溴仿 -
 溴乙烷
溴乙烷


參見
參考資料
- ^ 卤代烷 (PDF). 太平洋化工資源網: p.20. [2021-09-01]. (原始內容存檔 (PDF)於2021-08-31) (中文).
同一烴基鹵烷的相對密度:碘烷 > 溴烷 > 氯烷。
- ^ E. Cheng, J. Chow, Y.F. Chow ,A. Kai , S.L. Lee, W.H.Wong. HKDSE Chemistry A Modern View Second Edition. : 42.18. ISBN 9789888242948 (英語).
- ^ Dagani, M. J.; Barda, H. J.; Benya, T. J.; Sanders, D. C., Bromine Compounds, Ullmann's Encyclopedia of Industrial Chemistry, Weinheim: Wiley-VCH, 2005, doi:10.1002/14356007.a04_405
- ^ Bromomethane. PubChem. [2021-09-01]. (原始內容存檔於2013-06-19) (英語).
- ^ Gordon W. Gribble "The diversity of naturally occurring organobromine compounds" Chemical Society Reviews, 1999, volume 28, pages 335 – 346.doi:10.1039/a900201d
- ^ Rhoda A. Marshall, John T.G. Hamilton , M.J. Dring, D.B. Harper. Do vesicle cells of the red alga Asparagopsis (Falkenbergia stage) play a role in bromocarbon production? Chemosphere 52 (2003) 471–475.
- ^ Gordon, H. Laing. Sir James Young Simpson and Chloroform (1811-1870). The Minerva Group, Inc. 2002-11: 106–109 [11 November 2011]. ISBN 978-1-4102-0291-8. (原始內容存檔於2020-08-19).
- ^ 陳炳聖. 《萬物簡史》. 源樺. 2007. ISBN 986828421X.
- ^ Aug. Kekulé. Über die s. g. gepaarten Verbindungen und die Theorie der mehratomigen Radicale. Annalen der Chemie und Pharmacie. 1857, 104 (2): 129–150. doi:10.1002/jlac.18571040202.
- ^ CHAPTER 1 INTRODUCTION TO ORGANIC CHEMISTRY (PDF). 南伊利諾斯大學艾德華茲維爾分校. [2021-09-01]. (原始內容存檔 (PDF)於2020-10-30) (英語).
- ^ Additions to Alkenes: Regiochemistry. [2021-09-01]. (原始內容存檔於2020-02-19).
- ^ Illustrated Glossary of Organic Chemistry. 加利福尼亞大學洛杉磯分校. [2021-09-01]. (原始內容存檔於2021-09-01) (英語).
Peroxide effect: The change in regioselectivity of the addition of HBr to an alkene or alkyne in the presence of a peroxide. The regioselectivity for the addition reactions of other electrophiles such as HCl and H3O+ are not altered in the presence of a peroxide.
- ^ molbase. 2-溴丙烷. 上海摩庫數據技術有限公司. [2021-07-28]. (原始內容存檔於2021-07-28) (中文).
- ^ 有機化合物命名原則2017 (頁面存檔備份,存於網際網路檔案館). 中國化學會. [2018-1-17]
- ^ 卤代烷 (PDF). 太平洋化工資源網: p.5. [2021-08-31]. (原始內容存檔 (PDF)於2021-08-31) (中文).
把鹵烷看作是烷基和鹵素結合而成的化合物而命名,稱為某烷某鹵。
- ^ Dr. Ian Hunt,. Chapter 4: Alcohols and Alkyl Halides. [2021-07-28]. (原始內容存檔於2021-07-28) (英語).
Note that it contains three distinct type of steps
- ^ 國際純化學和應用化學聯合會,化學術語概略,第二版。(金皮書)(1997)。在線校正版: (2006–) "homolysis (homolytic)"。doi:10.1351/goldbook.H02851
- ^ E. Cheng, J. Chow, Y.F. Chow ,A. Kai , S.L. Lee, W.H.Wong. HKDSE Chemistry A Modern View Second Edition. : 44.4. ISBN 9789888242948 (英語).
- ^ THE REACTION BETWEEN SYMMETRICAL ALKENES AND BROMINE. 2000 [2021-07-28]. (原始內容存檔於2021-12-08) (英語).
- ^ Solomons, T.W. Graham; Fryhle, Craig B., Organic Chemistry 8th, Wiley, 2003, ISBN 0471417998
- ^ Smith, Janice G., Organic Chemistry 2nd, McGraw-Hill, 2007, ISBN 0073327492
- ^ P.J. Kropp, K.A. Dans, S.D. Crawford, M.W. Tubergen, K.D. Kepler, S.L Craig, and V.P. Wilson, Surface-mediated reactions. 1. Hydrohalogenation of alkenes and alkynes, J. Am. Chem. Soc., 1990, 112 (112): 7433–7434, doi:10.1021/ja00176a075.
- ^ Streiwieser, A.; Heathcock, C.H.; Kosower, E.M. 11.6.G. Alkenes: Reactions: Free Radical Additions. Introduction to Organic Chemistry 4th. New York: Macmillan. 1992: 288.
- ^ 邢其毅; 裴堅; 裴偉偉; 徐瑞秋. 醇羥基轉換爲鹵原子的反應. 基礎有機化學. 北京大學出版社. : 285 [2021-07-28]. ISBN 9787301272121 (中文).
- ^ 張永華, 丁辰元, 張國璽. 叔丁基溴合成方法的改進 (頁面存檔備份,存於網際網路檔案館)[J]. 首都師範大學學報:自然科學版, 1989(2):21-23.
- ^ 溴丙烷. molbase. 上海摩庫數據技術有限公司. [2021-07-28]. (原始內容存檔於2021-07-28) (中文).
另外,在紅磷存在下,正丙醇與溴反應也可製備溴丙烷。
- ^ Rolf Appel. Tertiary Phosphane/Tetrachloromethane, a Versatile Reagent for Chlorination, Dehydration, and P-N Linkage. Angewandte Chemie International Edition in English. 1975, 14 (12): 801–811. doi:10.1002/anie.197508011.
- ^ Cläre Hunsdiecker, et al. U.S. Patent # 2,176,181.
- ^ Heinz Hunsdiecker; Cläre Hunsdiecker. Über den Abbau der Salze aliphatischer Säuren durch Brom. Ber. 1942, 75: 291. doi:10.1002/cber.19420750309.
- ^ Borodin, A. Ueber Bromvaleriansäure und Brombuttersäure. Ann. 1861, 119: 121. doi:10.1002/jlac.18611190113.
- ^ Allen, C. F. H.; Wilson, C. V. (1955). "Methyl 5-bromovalerate". Org. Synth.; Coll. Vol. 3: 578.
- ^ Chakrabartty, in Trahanovsky, Oxidation in Organic Chemistry, pp 343-370, Academic Press, New York, 1978
- ^ March, Jerry; Smith, Michael B. Knipe, A.C. , 編. March's Advanced Organic Chemistry Reactions, Mechanisms, and Structure. 6th. Hoboken: John Wiley & Sons. 2007: 484. ISBN 9780470084946.
- ^ Reynold C. Fuson and Benton A. Bull. The Haloform Reaction. Chemical Reviews. 1934, 15 (3): 275–309. doi:10.1021/cr60052a001.
- ^ 8.4 Comparison and Competition Between SN1, SN2, E1 and E2. Pressbooks. [2021-07-28]. (原始內容存檔於2021-07-28) (英語).
- ^ SN1 and SN2 Reactions (pdf). [2021-07-28]. (原始內容存檔 (PDF)於2019-05-12) (英語).
- ^ 邢其毅; 裴堅; 裴偉偉; 徐瑞秋. 影響鹵代烴親核取代反應的因素. 基礎有機化學. 北京大學出版社. : 239 [2021-07-28]. ISBN 9787301272121 (中文).
- ^ Theory of Aetherification. Philosophical Magazine. 1850, 37: 350–356.)
- ^ Organikum, 22. Edition (German), Wiley-VCH, Weinheim, 2004, ISBN 3-527-31148-3
- ^ 羧酸的製備——腈水解 (頁面存檔備份,存於網際網路檔案館). 北京中醫藥大學遠程教育學院
- ^ Nishimura, Shigeo. Handbook of Heterogeneous Catalytic Hydrogenation for Organic Synthesis 1st. New York: Wiley-Interscience. 2001: 254–277. ISBN 9780471396987.
- ^ 42.0 42.1 March, Jerry (1985). Advanced Organic Chemistry, Reactions, Mechanisms and Structure, third Edition, John Wiley & Sons. ISBN 0-471-85472-7.
- ^ L. Friedman, Harold Shechter. Preparation of Nitriles from Halides and Sodium Cyanide. An Advantageous Nucleophilic Displacement in Dimethyl Sulfoxide. Journal of Organic Chemistry. 1960, 25: 877–879. doi:10.1021/jo01076a001.
- ^ Gabriel, S. Ueber eine Darstellung primärer Amine aus den entsprechenden Halogenverbindungen. Ber. 1887, 20: 2224 [2021-09-15]. (原始內容存檔於2020-04-10).
- ^ Sheehan, J. C.; Bolhofer, V. A. An Improved Procedure for the Condensation of Potassium Phthalimide with Organic Halides. J. Am. Chem. Soc. 1950, 72: 2786. doi:10.1021/ja01162a527.
- ^ Gibson, M.S.; Bradshaw, R.W. The Gabriel Synthesis of Primary Amines. Angew. Chem. Int. Ed. Engl. 1968, 7: 919. doi:10.1002/anie.196809191.
- ^ Mitsunobu, O. Comp. Org. Syn. 1991, 6, 79-85.(綜述)
- ^ Khan, M. N. Suggested Improvement in the Ing-Manske Procedure and Gabriel Synthesis of Primary Amines: Kinetic Study on Alkaline Hydrolysis of N-Phthaloylglycine and Acid Hydrolysis of N-(o-Carboxybenzoyl)glycine in Aqueous Organic Solvents. J. Org. Chem. 1996, 61, 8063-8068.
- ^ Stolz, Daniel; Kazmaier, Uli. Metal Enolates as Synthons in Organic Chemistry. PATai's Chemistry of Functional Groups. 2010. ISBN 9780470682531. doi:10.1002/9780470682531.pat0423.
- ^ Hart, David J.; Ha, Deok Chan. The ester enolate-imine condensation route to .beta.-lactams. Chemical Reviews. 1989, 89 (7): 1447–1465. doi:10.1021/cr00097a003.
- ^ Wu, George; Huang, Mingsheng. Organolithium Reagents in Pharmaceutical Asymmetric Processes. Chemical Reviews. 2006, 106 (7): 2596–2616. PMID 16836294. doi:10.1021/cr040694k.
- ^ Curti, Claudio; Battistini, Lucia; Sartori, Andrea; Zanardi, Franca. New Developments of the Principle of Vinylogy as Applied to π-Extended Enolate-Type Donor Systems. Chemical Reviews. 2020, 120 (5): 2448–2612. PMID 32040305. doi:10.1021/acs.chemrev.9b00481
 .
.
- ^ Alkylation of enolates. ChemistryScore. [2021-07-28]. (原始內容存檔於2021-07-28) (英語).
In the first step, the base (LDA) removes a proton from the α carbon to produce an enolate. In the second step, the enolate function as a nucleophile and attacks the alkyl halide, displacing the halide as a good leaving group. The alkylation product is formed by the SN2 reaction. This reaction works best with methyl and primary alkyl halides. Hindered alkyl halides and those with halides bonded to sp² hybridized carbon do not undergo substitution.
- ^ Smith, Janice Gorzynski. Organic Chemistry: Second Ed. 2008. pp 905-906
- ^ House, Herbert O. Modern Synthetic Reactions. Menlo Park, CA.: W. A. Benjamin. 1972. ISBN 0-8053-4501-9.
- ^ Malonic Ester Synthesis. Organic Chemistry Portal. [2007-10-26]. (原始內容存檔於2018-07-29).
- ^ Saytzeff, Alexander. Zur Kenntniss der Reihenfolge der Analgerung und Ausscheidung der Jodwasserstoffelemente in organischen Verbindungen. Justus Liebigs Annalen der Chemie. 1875, 179 (3): 296–301. doi:10.1002/jlac.18751790304.
- ^ David R. Klein. 有機化學天堂秘笈. Taiwan: 天下遠見出版股份有限公司. Oct 19, 2007: 302~309. ISBN 978-986-216-008-4 (中文(香港)).
- ^ Smith, M. B.; March, J. Advanced Organic Chemistry, 5th ed.; John-Wiley & Sons: New York, 2001. ISBN 0-471-58589-0 - 包含格氏試劑的若干反應。
- ^ Kürti, L.; Czakó, B. Storategic Applications of Named Reactions in Organic Chemistry; Elsevier: Amsterdam, 2005; pp 188–189. ISBN 0-12-429785-4
- ^ Stent, M. Generation of a Highly Basic and Nucleophilic Organolithium; Isopropyllithium. Synthetic Pages. 2002, (195) [2021-07-29]. (原始內容存檔於2016-03-10).
- ^ Modern Organocopper Chemistry, N. Krause Ed. Wiley-VCH, 2002.
- ^ J. F. Normant. Organocopper(I) Compounds and Organocuprates in Synthesis. Synthesis. 1972, 1972 (02): 63–80. doi:10.1055/s-1972-21833.
- ^ Adolphe Wurtz. Sur une nouvelle classe de radicaux organiques. Annales de chimie et de physique. 1855, 44: 275–312 [2021-07-28]. (原始內容存檔於2020-04-10).
- ^ March Advanced Organic Chemistry 5th edition p.535
- ^ Gary M. Lampman and James C. Aumiller "Bicyclo[1.1.0]butane" Organic Syntheses, 1971, volume 51, pp 55-9. doi:10.15227/orgsyn.051.0055 Aditya Krishna "Dihalides Quartz"
- ^ 邢其毅; 裴堅; 裴偉偉; 徐瑞秋. 鹵代烴與金屬的反應. 基礎有機化學. 北京大學出版社. : 265 [2021-07-28]. ISBN 9787301272121 (中文).
- ^ Johnson, J. Enoch; Blizzard, Ronald H.; Carhart, Homer W. Hydrogenolysis of alkyl halides by lithium aluminum hydride.. Journal of the American Chemical Society. 1948, 70 (11): 3664. PMID 18121883. doi:10.1021/ja01191a035.
- ^ Krishnamurthy, S.; Brown, Herbert C. Selective reductions. 28. The fast reaction of lithium aluminum hydride with alkyl halides in THF. A reappraisal of the scope of the reaction. The Journal of Organic Chemistry. 1982, 47: 276. doi:10.1021/jo00341a018.
- ^ 邢其毅; 裴堅; 裴偉偉; 徐瑞秋. 鹵代烴的還原. 基礎有機化學. 北京大學出版社. : 259 [2021-07-28]. ISBN 9787301272121 (中文).
- ^ Carruthers, W. Some modern methods of organic synthesis. Cambridge University Press. 2004: 470. ISBN 0521311179.
- ^ 邢其毅; 裴堅; 裴偉偉; 徐瑞秋. 鹵代烴親核取代反應的應用. 基礎有機化學. 北京大學出版社. : 248-249 [2021-07-28]. ISBN 9787301272121 (中文).
- ^ 卤烷的物理性质 (PDF). 太平洋化工資源網: p.20. [2021-09-01]. (原始內容存檔 (PDF)於2021-08-31) (中文).
- ^ 三溴甲烷. chemicalbook. [2021-07-28]. (原始內容存檔於2021-07-28) (中文).
另外可作為消毒劑、鎮痛劑、致冷劑、防火化學品,測定分子量的溶劑以及有機合成的中間體。
- ^ 溴乙烷. chemicalbook. [2021-07-28]. (原始內容存檔於2021-07-28).
用於醫藥、農藥、染料工業,是有機合成中的乙基化劑,也用作致冷劑和有機溶劑